

This is the Chapter 11 of Py4Bio.
Biopython is a library for the Python programming language.
Package that assists with processing biological data.
Consists of several modules – some with common operations, some more specialized.
But before learning more about BioPython, let’s get some idea about Object Oriented Programming (OOP).
Biopython is object-oriented. So, some knowledge helps understand how biopython work.
Object Oriented Programming
OOP is a way of organizing data and methods that work on them in a coherent package. OOP helps structure and organize the code.
Classes and objects
A class:
- is a user defined
type - is a mold for creating objects
- specifies how an object can contain and process data
- represents an abstraction or a template for how an object of that class will behave
An object is an instance of a class.
All objects have a type – shows which class they were made from.
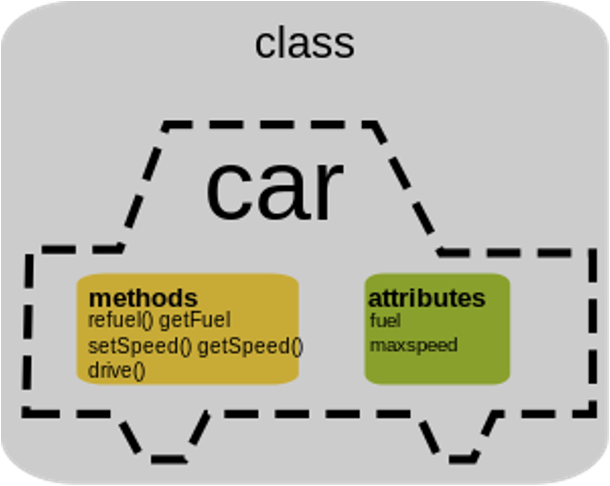
Attributes and methods
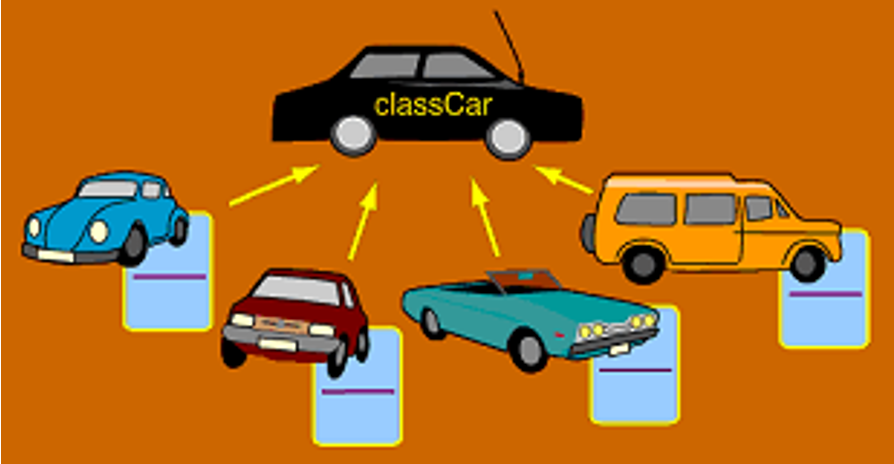
Class and Instances
Class and object example
Class: Seq
Seq has:
- attribute length
- method translate
An object of the class Seq is created like this:
myseq = Seq(“ATGGCCG”)Get sequence length:
myseq.lengthGet sequence translation:
myseq.translate()OOP Summary
An object has to be instantiated, i.e. created, to exist.
Every object has a certain type, i.e. is of a certain class.
The class decides which attributes and methods an object has.
Attributes and methods are accessed using . after the object variable name.
Explaining OOP with BioPython
You can install BioPython from here.
With time, you will come to know many other packages. It’s a good idea to familiarize yourself with the documentation of that package. For example, BioPython documentation can be found here.
Test your BioPython installation:
>>> import Bio
>>> print(Bio.__version__)Biopython functionality and tools
Tools to parse bioinformatics files into Python data structures
Supports the following formats
- BLAST, Clustalw, FASTA
- PubMed and Medline
- ExPASy files
- SwissProt, PDB
Files in the supported formats can be iterated over record by record or indexed and accessed via a dictionary interface.
Seq object
Represents one sequence. It has following methods:
- translate()
- transcribe()
- complement()
- reverse_complement()
>>> from Bio.Seq import Seq
>>> my_seq = Seq("AGTACACTGGT")
>>> my_seq
Seq('AGTACACTGGT')
>>> my_seq.translate()
Seq('STL')
>>> my_seq.transcribe()
Seq('AGUACACUGGU')
>>> my_seq.complement()
Seq('TCATGTGACCA')
What is happening here?
from Bio.Seq import SeqBio and Seq are packages.
Packages contain modules.
We are importing Seq module from the Seq package.
Modules are essentially individual Python file.
Seq module contains class Seq()
We are making an instance of Seq() class in my_seq variable.
translate, transcribe, complements are methods defined in Seq() class.
We are calling those methods for my_seq instance we just created.
Few more examples with Seq
All transcribe() does is a switch T –> U. lThe Seq object also includes a back-transcription method.
from Bio.Seq import Seq
coding_dna = Seq("ATGGCCATTGTAATGGGCCGCTGAAAGGGTGCCCGATAG")
messenger_rna = coding_dna.transcribe()
cDNA = messenger_rna.back_transcribe()
print(coding_dna)
print(messenger_rna)
print(cDNA)Seq as a string
Most string methods work on Seqs
If string is needed, do str(seq)
>>> seq = Seq('CCGGGTTAACGTA')
>>> seq[:5]
Seq('CCGGG', IUPACUnambiguousDNA())
>>> len(seq)
13
>>> seq.lower()
Seq('ccgggttaacgta', DNAAlphabet())
>>> print(seq)
CCGGGTTAACGTA
>>> list(seq)
['C', 'C', 'G', 'G', 'G', 'T', 'T', 'A', 'A', 'C', 'G', 'T', 'A']
>>> mystring = str(seq)
>>> print(mystring)
CCGGGTTAACGTA
>>> type(seq)
<class 'Bio.Seq.Seq'>
>>> type(mystring)
<type 'str'>Introduction to SeqRecord
Seq contains the sequence. But sequences often come with a lot more.
SeqRecord = Seq + metadata
Main attributes: ID and Seq
But it may have additional attributes:
- name – Sequence name, e.g. gene name (string)
- description – Additional text, imagine fasta description (string)
- dbxrefs – List of database cross references (list of strings)
- features – Any (sub)features defined (list of SeqFeature objects)
- annotations – Further information about the whole sequence (dictionary) Most entries are strings, or lists of strings.
from Bio.SeqRecord import SeqRecord
from Bio.Seq import Seq
seq = Seq('CCGGGTTAACGTA')
seq_record_1 = SeqRecord(seq, id='01')
print(seq_record_1)
# Or,
seq_record_1 = SeqRecord(seq)
seq_record_1.id = "01"
seq_record_1.descrption = "toxic membrane protein"
print(seq_record_1)
# Or,
#another way to define a Seq Record
seq_record_1 =
SeqRecord(Seq('CCGGGTTAACGTA'), id = 'YP_025292.1', name='HokC', description='toxic membrane protein',dbxrefs=[])
print(seq_record_1)SeqIO: Another important module of BioPython
We will never type sequence in a program. We’ll read it from a file in different format (fasta, GenBank, etc.).
SeqIO provides tools to to retrieve sequences as SeqRecord, and can write SeqRecord to file.
Reading: parse(file_handle, format)
Writing: write(SeqRecords(s), file_handle, format)
Read/write common biological file formats:
- FASTA, GenBank, EMBL
- FASTQ (sequencing reads)
- GFF, BED (genomic features)
- PDB (protein structures)
- PHYLIP, Nexus (phylogenetic data)
from Bio import SeqIOSeqIO.parse()
Reads in sequence data as SeqRecord objects
It expects two arguments:
An object (called handle) to read the data. It can be:
- a file opened for reading
- the output from a command line program
- data downloaded from the internet
A lower case string specifying the sequence format
The object returned by SeqIO.parse() is an iterator which returns SeqRecord objects.
from Bio import SeqIO
file_handle = "P53.fasta" # notice we are not using open()
for seq_rec in SeqIO.parse(file_handle, "fasta"):
print(seq_rec.id)
print(seq_rec.seq)
print(len(seq_rec.seq))Writing files
from Bio import SeqIO
seq_list = [ ... ] # this should be a list of SeqRecords
SeqIO.write(seq_list, "seq_list.fasta", "fasta")Example Use Case: Fetching a sequence from NCBI
from Bio import Entrez, SeqIO
Entrez.email = "your@email.com"
handle = Entrez.efetch(db="nucleotide",
id="NM_001301717", rettype="gb", retmode="text")
record = SeqIO.read(handle, "genbank")
print(record.description)Exercise 1: Filtering DNA sequences
Input data: DNA sequencing reads in FASTQ format in file sample1.fq
Problem: Write a script that:
1. reads the sequences
2. writes only those reads which
a) are >= 200 bp long
b) do not contain any uncalled bases (i.e. ‘N’ s) to an output file sample1_filtered.fq
Exercise 2: Calculate FASTQ stats
Input data: DNA sequences in FASTQ format from file reads.fq
Problem: Write a script that
a) calculates the following values for each read:
id, length, GC content, A count, T count, C count, G count, avg. quality
b) writes these as columns to a csv file (one row per read)
Hints:
– You can use Bio.SeqUtils for calculating GC content and skew (check documentation!)
– Find base qualities for each record in record.letter_annotations[“phred_quality”]
Go to the index of Py4Bio.
Leave a Reply