

Once we assembled and annotated bacterial genomes, next step after inferring phylogeny usually is to finding and clustering orthologous genes.
What are orthologs? Well, Genetics 101 tells us that they are genes evolutionary shared between multiple taxa descended from common ancestors. That means a series of (pretty-much) reliable replications and cell division passed between these set of genes and their common ancestor. They are not duplicates (or paralogs) and they have been mostly vertically transmitted.
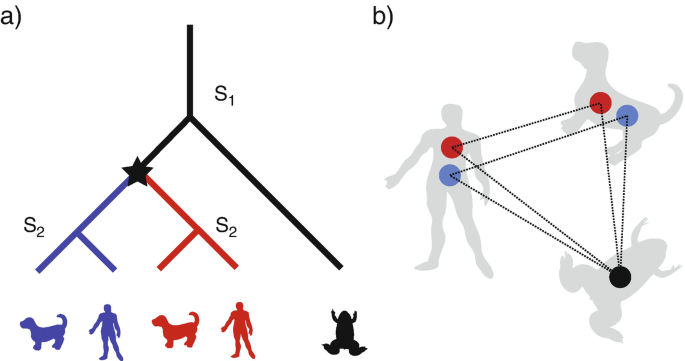
Why orthologs are important? Well, ortholog gene clusters has many practical uses. For example, if we can successfully identify set of conserved orthologs between multiple taxa, we can get the “core” of the species chromosome, i.e. set of genes that pretty much define the species we are working with. The phylogeny constructed from the “core” genome will give us a very reliable history of the bacterial species in question.
We can also use the ortholog groups to infer individual gene tree, estimate evolutionary pressure across different parts of the genome, define the accessory and unique genes (pan-genome analysis). Even going further, if we already have characterized the bacterial strains for some traits, we can do GWAS analysis based on the (orthologous) gene present-absent matrix and can potentially find genes associated with the trait!
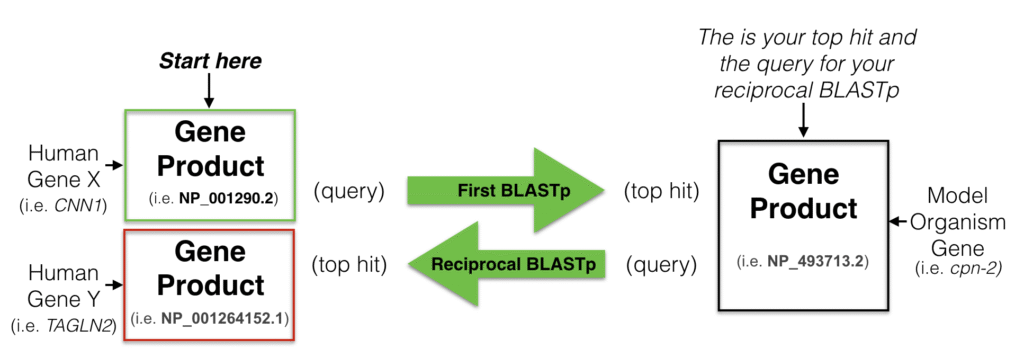
Now, hopefully I’ve made you excited enough to jump into doing an ortholog analysis. The question is, how do we do it? The algorithm to finding genes that share ortholog-relationship is simple. We just do two-way homology search (BLAST), i.e. taking a homology-search hit as a query to check if the previous query is also a homology-search hit or not. If they are, they are (probably) coming from the same ancestor.
However, the devil is always in the details. For example, what similarity cut-off you will be using to determine if two y are orthologs? If you find a set of genes from different taxa with some kind of two-way similarity, how do you “group” them together, i.e. which clustering algorithm to use?
There are so many different ortholog finding programs available and they approach the problems using differently. In this post, we will use PIRATE.
PIRATE actually uses multiple similarity-threshold cutoffs, since different gene-family may have different level of homology between them. Then it uses MCL clustering to determine the clusters, which does a great job to identify the clusters.
In this tutorial I’ll show you how to do ortholog clustering from assembled genomes.
Dataset description
We will use 44 genomes of Propionibacterium freudenreichii isolates coming from yogurt samples.
Directory structure will look like following:
- casestudy_lactobacillus
|- annotation
|- pirate
|- input
|- input_fixed
|- outputYou can download the genomes from NCBI using BioProject:
mkdir casestudy_lactobacillus
cd casestudy_lactobacillus
# Download 44 genomes
datasets download genome accession PRJNA946676
unzip ncbi_dataset.zip
# Copying the fasta file in a genomes folder
mkdir annotation
cd annotation
mv ../ncbi_dataset/data/GCA*/*.fna .
# Cleaning NCBI files
cd ..
rm -rf md5sum.txt ncbi_dataset* README.mdGenome annotation
PIRATE needs annotation file in GFF format. You can use Bakta. For this tutorial, I will be using Beav pipelin. Beav uses Bakta to annotate bacterial genome, and then uses many other tools on top of it to annotate other interesting features. The GFF files can be downloaded from here.
Prepare GFF for PIRATE run
I’m assuming you have GFF annotation files in the annotation directory. Let’s put them together:
mkdir pirate
cd pirate
mkdir input
cd input
ln -s ~/path/to/casestudy_lactobacillus/annotation/*/*_final.gff3 .Before we start running PIRATE for ortohlogous gene clustering, we need to make sure that the different feature names in the GFF files are consistent. We can use AGAT‘s agat_convert_sp_gxf2gxf.pl tool which can removes duplicate features, fixes duplicated IDs, adds missing ID.
We also want to add the genome sequence at the end of GFF file, so that PIRATE can also extract the sequences.
Here’s the AGAT script I use:
#!/bin/bash
for infile in `ls -1 input/*final.gff3`; do
strain=`echo -e "$infile" | sed 's/^.*\///g;s/.gff3//g' | sed 's/_final//g' `
echo -e "$strain"
agat_convert_sp_gxf2gxf.pl -g $infile -o ${strain}.gff --gff_version_output 3
echo -e "##FASTA" >> ${strain}.gff
cat ~/path/to/casestudy_lactobacillus/annotation/${strain}.f* >> ${strain}.gff
doneNow, create a new directory for updated GFF files and move to PIRATE run.
mkdir input_fixed log
mv *gff input_fixed
mv *agat.log logYou can run PIRATE using following command:
PIRATE -i input_fixed/ -o output/ -t 16This will take some time. PIRATE will construct pangenome, do BLAST, do MCL clustering, and finalize the orthologous and paralogous clusters. You should check the PIRATE.log file. Pirate ends with a joke, for example:
Q: What's a pirate's favourite programming language?
A: R. Although there are those that say his true love be the C.
YARR!
Generate gene presence-absence matrix
Pirate will create PIRATE.gene_families.ordered.tsv file that defines all the gene clusters, annotation information, and genome-location they are present in. However, this file is not super user friendly. PIRATE comes with few tools which are located in the installation folder. We can run the these tools to address this issue.
(You can also download these scripts from PIRATE repo)
# Rename with original locus tag form input files
subsample_outputs.pl -i output/PIRATE.gene_families.ordered.tsv -g output/modified_gffs/ -o output/PIRATE.gene_families.ordered.originalIDs.tsv --field "prev_ID" --feature "CDS"
# Gene/allele presence-absence
PIRATE_to_Rtab.pl -i output/PIRATE.gene_families.ordered.originalIDs.tsv -o output/PIRATE.gene_families.ordered.originalIDs.genePA.tsv
# Paralog presence-absence (duplications = d, fission/fusions = ff)
paralogs_to_Rtab.pl -i output/PIRATE.gene_families.ordered.originalIDs.tsv -o output/PIRATE.gene_families.ordered.originalIDs.genePAparalogs.tsv --type d
# Create roary format output
PIRATE_to_roary.pl -i output/PIRATE.gene_families.ordered.originalIDs.tsv -o output/PIRATE.gene_families.ordered.originalIDs.roary.tsv
# Extract all sequences for each ortholog group
align_feature_sequences_mod.pl -i output/PIRATE.gene_families.ordered.tsv -f -o output/all_sequences -g output/modified_gffs/ -a
Once we have the PIRATE analysis done, we can create a core-genome phylogeny, do a pan-genome analysis, do microbial GWAS, extract gene of interests, evolutionary selection analysis. The sky is the limit here!
I will write detail tutorial for each of these analysis using real-life datasets that you can reproduce. Subscribe to get email notification of new updates, and spread the word!
Leave a Reply