
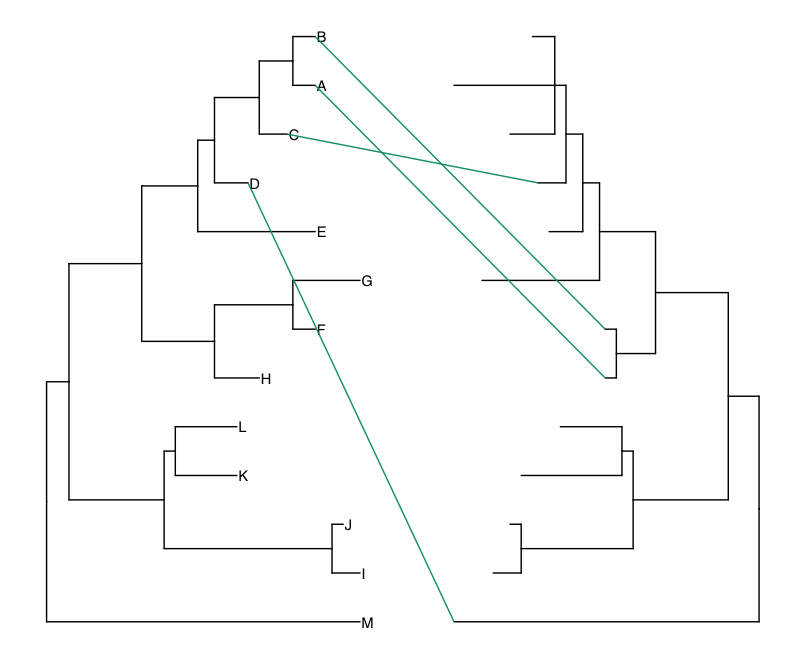
Tanglegram is a representation of co-phylogeny where two phylogenetic trees are linked. This method is super useful to visualize common traits shared by both trees. For example, it can be used to visualize host-pathogen (or host-symbiotic) evolution and visualize if there is any phylogenetic concordance between the two phylogenetic trees.
I was in need to visualize co-phylogeny of phylogenetic tree reconstructed from chromosomal and symbiotic genes. Surprisingly, I didn’t find any straight-forward solution in R that can be used for drawing tanglegram. Particularly I wanted to leverage the beautiful ggtree library. After trying out several methods, I found the following approach works well for me so far. I have released a small R package on it, which can be found on GitHub.
How to make a Tanglegram from scratch
In this post, I’m going to use two toy trees with the following Newick format. Note that they have the same isolate, but different tree-topology (since supposedly different gene-set were used to reconstruct them).
The datasets used in this tutorial can be downloaded from here.
Tree 1: (((((((A:4,B:4):6,C:5):8,D:6):3,E:21):10,((F:4,G:12):14,H:8):13):13,((I:5,J:2):30,(K:11,L:11):2):17):4,M:56);
Tree 2: (((((((F:8,I:18,G:4):2,C:5):3,M:6):3,E:21):10,((A:2,B:2):4):3):13,((K:5,L:2):20,(H:18,J:11):2):17):4,D:56);We might be interested to visualize one (or more) interesting feature(s) (i.e. genotype) associated with the isolates in both trees. Our meta-file looks like the following:
| Isolate | Genotype |
| A | Green |
| B | Green |
| C | Green |
| D | Green |
| E | Red |
| F | Red |
Let’s load the necessary packages.
library(ggplot2)
library(ggtree)
library(phangorn)
library(dplyr)The best thing about ggtree is you can attach any feature(s) associated with isolates using a simple CSV file. Just make sure that the first column of your metafile has the same isolate names as used in the tree. Let’s load the metafile and both phylogenetic tree to be used. I also like to do midpoint rooting at this point.
# Meta file
meta <- read.table('~/path/to/meta.csv', sep=',', header = T)
# Load tree 1
tree1 <- read.tree('~/path/to/tree1.nwk')
tree1 <- midpoint(tree1)
# Load tree 2
tree2 <- read.tree('~/path/to/tree2.nwk')
tree2 <- midpoint(tree2)Let’s combine the meta feature dataset with both phylogenetic trees and visualize how they look.
t1 <-ggtree(tree1) %<+% meta + geom_tiplab()
t2 <- ggtree(tree2) %<+% meta + geom_tiplab()
t1
t2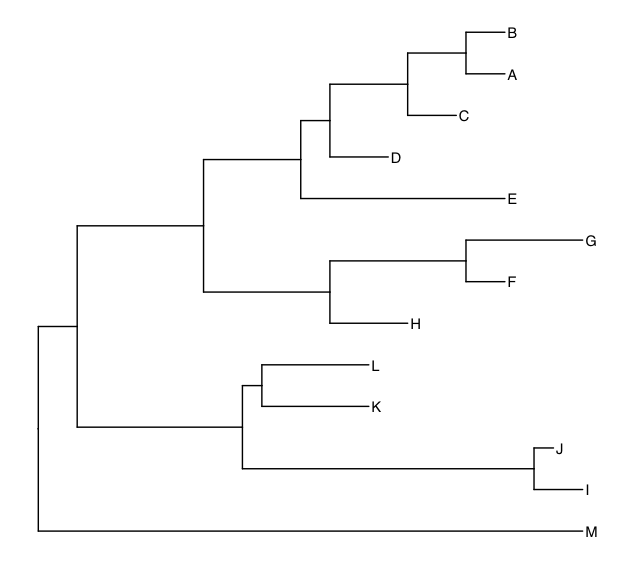

Now we are going to draw both trees in a single figure. We also want to flip tree 2, for which we need to change the x-coordinates in that tree.
d1 <- t1$data
d2 <- t2$data
d1$tree <-'t1'
d2$tree <-'t2'
d2$x <- max(d2$x) - d2$x + max(d1$x) + max(d1$x)*0.3
pp <- t1 + geom_tree(data=d2)
pp In the above code block, we are grabbing the backend data frame from both trees and updating the tree 2 data frame x-coordinate. We are using this equation for the update: max(d2$x) - d2$x + max(d1$x) + max(d1$x)*0.3. You can toy with different values depending on the branch length unit of your tree to get good visualization (I particularly suggest changing max(d1$x)*0.3 terms).

Let’s join d1 and d2 for dataset so that we can use the coordinates of the tips for making connections between both of the trees.
dd <- bind_rows(d1, d2) %>%
filter(isTip == TRUE)
dd1 <- as.data.frame(dd) Now, we are going to conditionally join the tips of both trees for the feature we are interested in. Connected tips will represent the same isolates.
green_tree <- dd1[which(dd1$Genotype == 'Green'), c('label', 'x', 'y', 'tree')]
pp + geom_line(aes(x, y, group=label), data=green_tree, color='#009E73') 
Connecting the isolates
Lily asked in the comment section if it is possible to connect all the tip labels from both trees. The previous code chunk actually connects the tips based on a subset from meta. In this case, we do not have to do any kind of subsetting. Using the following code we can connect all tips:
pp + geom_line(aes(x, y, group=label), data=dd1)Here, label is the tip-label variable, which is already associated with cophylogeny. You can check it by head(pp$dd1)

This may look messy since I am using totally random trees. However, if your co-phylogeny trees have some particular pattern, the tanglegram will show that.
Tangler: The R package
Due to the popularity of this tutorial, I have released a small R package that can help you to draw simple tanglegram from two ggtree objects.
The R package called TangleR, currently released in GitHub.
You can download it in R using the following command:
library("devtools")
install_github('acarafat/tangler')Here’s how to use this TangleR package:
library(ggtree)
library(tangler)
library(ggnewscale)
library(dplyr)
library(ggplot2)
# Load trees
t1 <- read.tree('data/tree1.nwk')
t2 <- read.tree('data/tree2.nwk')
# Load meta
meta=read.csv('tree_meta.csv', header=T)
# Annotate trees
tree1 <- ggtree(t1) %<+% meta +
geom_tiplab() +
geom_tippoint(aes(color=Genotype))
tree2 <- ggtree(t2) %<+% meta + geom_tiplab()
# Draw Tanglegram
simple.tanglegram(tree1, tree2, Genotype, Green, tiplab = T)
# Update the connecting line x-position so that it do not overlap with tip-labels.
simple.tanglegram(tree1, tree2, Genotype, Green, l_color = 'green3', t2_pad = 0.3,
tiplab = T, lab_pad = 0.1, x_hjust = 1, t2_tiplab_size = 3)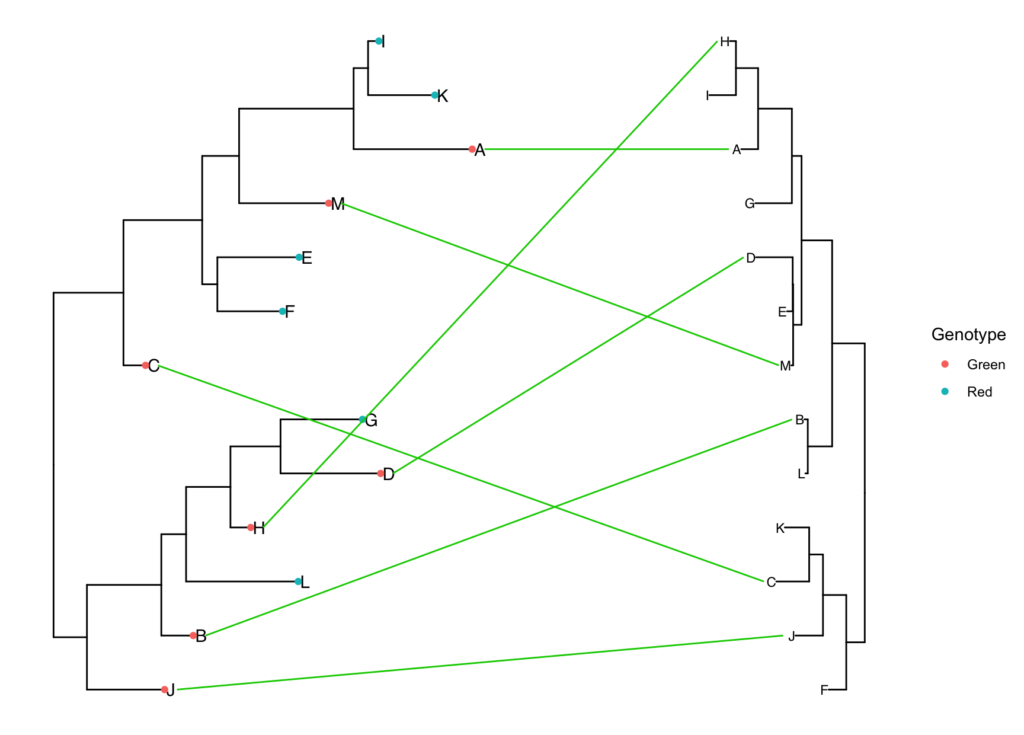
Now let’s say you want to reorder the tips of both phylogeny so that the tips are better aligned, however the overall tree toplogy is unchanged. For that you can use the pre.rotate function. The only difference is, you need to make sure that you are using ladderize=FALSE when converting the rotated tree in a ggtree object, otherwise ggtree will override tip-order.
# Rotate the internal nodes so that tips of both trees are aligned
rotated_trees <- pre.rotate(t1, t2)
t1 <- rotated_trees[[1]]
t2 <- rotated_trees[[2]]
# Annotate Trees, make sure to set ladderize=F
tree1 <- ggtree(t1, ladderize=F) %<+% meta +
geom_tiplab() +
geom_tippoint(aes(color=Genotype))
# Annotate Tree 2
tree2 <- ggtree(t2, ladderize=F) %<+% meta + geom_tiplab()
# Tanglegram, no line color
simple.tanglegram(tree1, tree2, Genotype, l_color = 'green3', Green, t2_pad = 0.3,
tiplab = T, lab_pad = 0.1, x_hjust = 1, t2_tiplab_size = 3)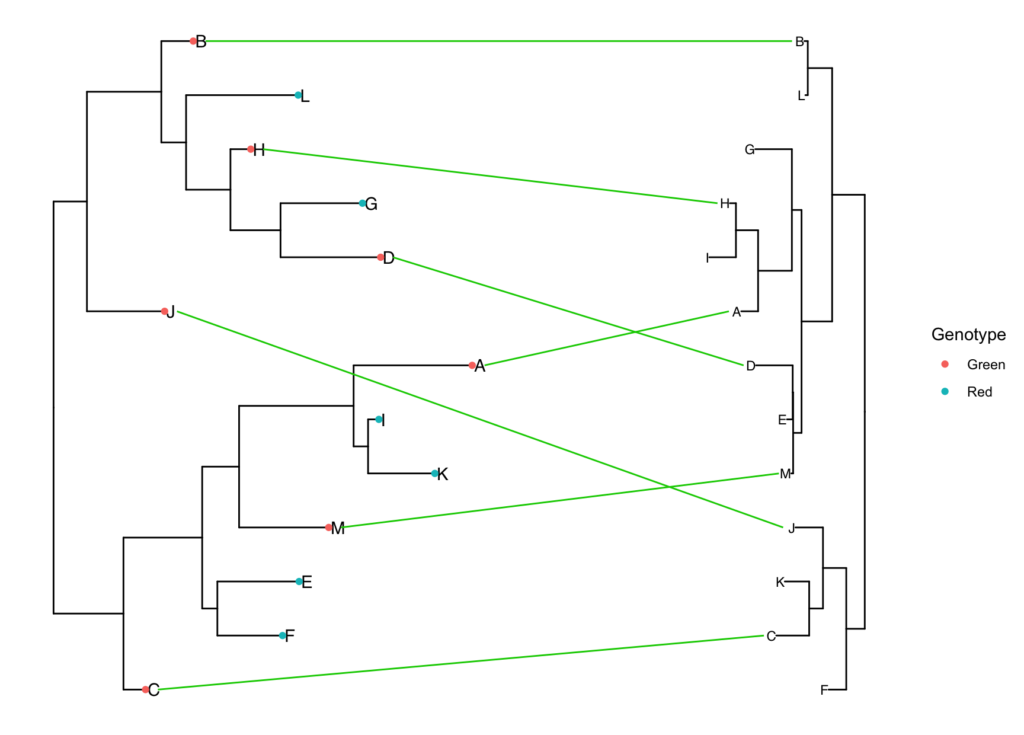
You can also draw tanglegram for all traits in the column using common.tanglegram function.
common.tanglegram(tree1, tree2, column = 'Genotype', sampletypecolors = c('green4', 'red'), t2_pad = 0.3,
tiplab = T, lab_pad = 0.1, t2_tiplab_size = 3)
Leave a Reply